
But the preprint culture is nascent in many areas of science (e.g., biology), and it seems to me that some journals are not yet fully comfortable with the idea. I was pleasantly surprised to learn, just in the last day or two, that most journals now have official policies that allow online distribution of preprints prior to publication. (This has been the case in theoretical physics since before I entered the field!) Let's hope that progress continues.
The work presented below applies ideas from compressed sensing, L1 penalized regression, etc. to genomic prediction. We exploit the phase transition behavior of the LASSO algorithm to construct a good genomic predictor for human height. The results are significant for the following reasons:
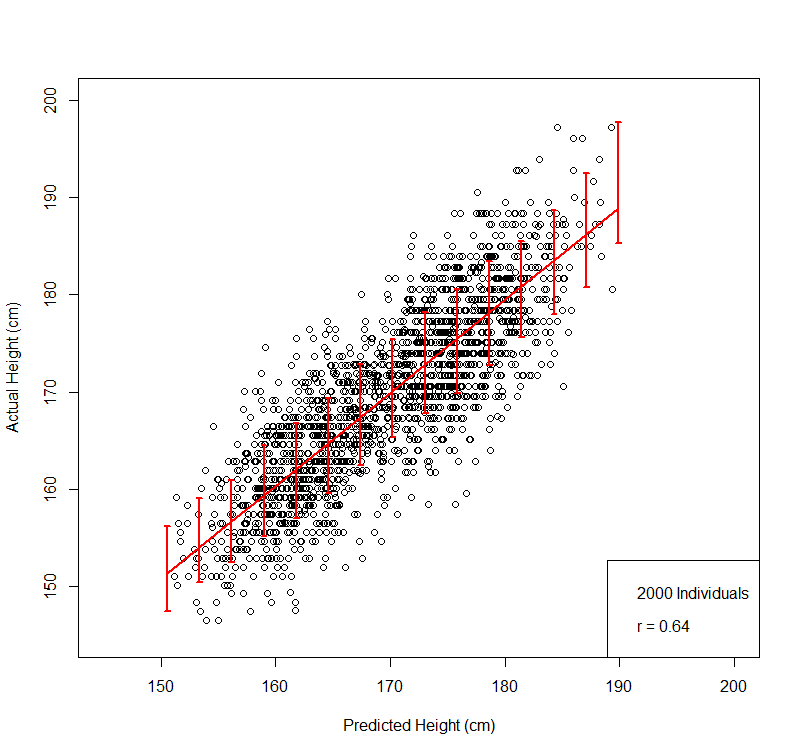
We applied novel machine learning methods ("compressed sensing") to ~500k genomes from UK Biobank, resulting in an accurate predictor for human height which uses information from thousands of SNPs.The scatterplot figure below gives an immediate feel for the accuracy of the predictor.
1. The actual heights of most individuals in our replication tests are within a few cm of their predicted height.
2. The variance captured by the predictor is similar to the estimated GCTA-GREML SNP heritability. Thus, our results resolve the missing heritability problem for common SNPs.
3. Out-of-sample validation on ARIC individuals (a US cohort) shows the predictor works on that population as well. The SNPs activated in the predictor overlap with previous GWAS hits from GIANT.
Accurate Genomic Prediction Of Human HeightThis figure compares predicted and actual height on a validation set of 2000 individuals not used in training: males + females, actual heights (vertical axis) uncorrected for gender. For training we z-score by gender and age (due to Flynn Effect for height). We have also tested validity on a population of US individuals (i.e., out of sample; not from UKBB).
(bioRxiv)
Louis Lello, Steven G. Avery, Laurent Tellier, Ana I. Vazquez, Gustavo de los Campos, and Stephen D.H. Hsu
We construct genomic predictors for heritable and extremely complex human quantitative traits (height, heel bone density, and educational attainment) using modern methods in high dimensional statistics (i.e., machine learning). Replication tests show that these predictors capture, respectively, ∼40, 20, and 9 percent of total variance for the three traits. For example, predicted heights correlate ∼0.65 with actual height; actual heights of most individuals in validation samples are within a few cm of the prediction. The variance captured for height is comparable to the estimated SNP heritability from GCTA (GREML) analysis, and seems to be close to its asymptotic value (i.e., as sample size goes to infinity), suggesting that we have captured most of the heritability for the SNPs used. Thus, our results resolve the common SNP portion of the “missing heritability” problem – i.e., the gap between prediction R-squared and SNP heritability. The ∼20k activated SNPs in our height predictor reveal the genetic architecture of human height, at least for common SNPs. Our primary dataset is the UK Biobank cohort, comprised of almost 500k individual genotypes with multiple phenotypes. We also use other datasets and SNPs found in earlier GWAS for out-of-sample validation of our results.
This figure illustrates the phase transition behavior at fixed sample size n and varying penalization lambda.

These are the SNPs activated in the predictor -- about 20k in total, uniformly distributed across all chromosomes; vertical axis is effect size of minor allele:

The big picture implication is that heritable complex traits controlled by thousands of genetic loci can, with enough data and analysis, be predicted from DNA. I expect that with good genotype | phenotype data from a million individuals we could achieve similar success with cognitive ability. We've also analyzed the sample size requirements for disease risk prediction, and they are similar (i.e., ~100 times sparsity of the effects vector; so ~100k cases + controls for a condition affected by ~1000 loci).
Note Added: Further comments in response to various questions about the paper.
1) We have tested the predictor on other ethnic groups and there is an (expected) decrease in correlation that is roughly proportional to the "genetic distance" between the test population and the white/British training population. This is likely due to different LD structure (SNP correlations) in different populations. A SNP which tags the true causal genetic variation in the Euro population may not be a good tag in, e.g., the Chinese population. We may report more on this in the future. Note, despite the reduction in power our predictor still captures more height variance than any other existing model for S. Asians, Chinese, Africans, etc.
2) We did not explore the biology of the activated SNPs because that is not our expertise. GWAS hits found by SSGAC, GIANT, etc. have already been connected to biological processes such as neuronal growth, bone development, etc. Plenty of follow up work remains to be done on the SNPs we discovered.
3) Our initial reduction of candidate SNPs to the top 50k or 100k is simply to save computational resources. The L1 algorithms can handle much larger values of p, but keeping all of those SNPs in the calculation is extremely expensive in CPU time, memory, etc. We tested computational cost vs benefit in improved prediction from including more (>100k) candidate SNPs in the initial cut but found it unfavorable. (Note, we also had a reasonable prior that ~10k SNPs would capture most of the predictive power.)
4) We will have more to say about nonlinear effects, additional out-of-sample tests, other phenotypes, etc. in future work.
5) Perhaps most importantly, we have a useful theoretical framework (compressed sensing) within which to think about complex trait prediction. We can make quantitative estimates for the sample size required to "solve" a particular trait.
I leave you with some remarks from Francis Crick:
Crick had to adjust from the "elegance and deep simplicity" of physics to the "elaborate chemical mechanisms that natural selection had evolved over billions of years." He described this transition as, "almost as if one had to be born again." According to Crick, the experience of learning physics had taught him something important — hubris — and the conviction that since physics was already a success, great advances should also be possible in other sciences such as biology. Crick felt that this attitude encouraged him to be more daring than typical biologists who tended to concern themselves with the daunting problems of biology and not the past successes of physics.
No comments:
Post a Comment